By R. G. MARQUART and I.
KATSNELSON, Fein-Marquart Associates, Baltimore, MD 21212, USA, G. W. A. MILNE,
National Institutes of Health, Bethesda, MD 20014, USA, S. R. HELLER, Environmental
Protection Agency, Washington, DC 20460, USA, G. G. JOHNSON JR, Computer Science
Department, Pennsylvania State University, University Park, PA 16802, USA and R. J.
JENKINS, Philips Electronic Instruments, Inc., 85 McKee Drive, Mahwah, NJ 07430, USA
A computer program which can identify the crystalline component phases of a mixture from its
X-ray diffraction pattern is described. The program uses the data base of powder diffraction
patterns maintained by the Joint Committee on Powder Diffraction Standards. The 'reverse
searching' technique employed permits the identification of each component and provides an
estimate of confidence in the identification. Errors in measured spacings and intensities are
tolerated by the program. The interactive search-match system is available for general use via a
network timeshared computer.
The NIH-EPA Chemical Information System (CIS) (Heller, Milne & Feldmann, 1977) is a collection of numeric databases together with a battery of computer programs that permit interactive, on-line searching through these data bases with the express purpose of identifying chemical substances from their measured physical properties. The types of analytical data that are used in this way by the CIS include mass spectra (Heller et al., 1976; Heller, Milne & Feldmann, 1976) and C nuclear magnetic resonance spectra (Dalrymple, Wilkins, Milne & Heller, 1978). Much of the focus of CIS development has been upon the identification of organic compounds - a task for which mass and NMR spectra are well suited. Since the passage in 1976 of the Toxic Substances Control Act*, however, it has become clear that any useful chemical information system must provide the capability of identifying inorganic as well as organic chemical substances.
Consequently, discussions between the management of the CIS and the joint Committee for Powder Diffraction Standards (JCPDS)** were initiated in 1976 with a view to defining the terms upon which the X-ray powder diffraction data base maintained by the Joint Committee might be merged into the CIS where it would be searched by CIS programs and provide a means for facile identification of substances from their powder diffraction patterns.
Computer programs for searching and matching files of X-ray powder diffraction data have been published (Frevel & Adams, 1968; Frevel, Adams & Ruhberg, 1976; Nichols, 1966; Johnson, 1977) but these had in common the disadvantages that they were batch programs, and were relatively difficult and expensive to use. There existed, therefore, a clear need for an efficient and cost-effective algorithm that could handle mixed patterns and the CIS undertook the task of writing such a program. The result of this work is the search-match system described in this paper. The program, using the current data base of the JCPDS, is now available on a fee-for-service basis via a commercial computer network (Heller, Milne & Feldmann, 1977) as part of the CIS for low-cost searching.
Material that can be identify by powder diffraction methods are often mixtures of inorganic compounds. This causes a problem in that the so-called "forward Searching" techniques, which are designed for use with pure compounds, do not work well with the data derived from mixtures. The identical problem arises in mass spectrometry, where "mixed spectra" may also be obtained. A solution that has been applied to the mass spectrometric case is that of reverse searching (Abramson, 1975), an example of which is to be found in the probability-based matching algorithms devised by McLafferty's group (Pesyna, Venkataraghavan, Dayringer & McLafferty, 1976). Reverse search algorithms compare each library pattern, one by one, to that of the unknown. When a match is found and accepted, that entire pattern can be mathematically removed from the pattern of the unknown and the search process re-initiated in an attempt to identify the remaining components of the mixture.
A complete Powder Diffraction Search-Match system (PSDM), based upon reverse
searching, has been designed, written and implemented. The purpose of this paper is to describe
this program and to present and discuss results obtained with it.
In the reverse search approach, the unknown pattern is not used as a source of searchable features for interrogation of the data base, as in normal, or "forward"searching. Rather, each pattern in the data base is considered as a possible match, and is compared to the data corresponding to the unknown to see if it might be contained within it.
This kind of search can be extremely time-consuming and expensive unless each of its
component steps is optimized. In PSDM, this optimization begins with the preparation from the
basic JCPDS file of a search file that contains only that information essential to the pattern-matching process. These data are then organized so as to minimize both the time required for
access and the computation once the search records have been retrieved.
(a) Selection of lines for use in the search programs
Principal among these system parameters was the number of lines from each library pattern to be retained in the search file. Other reverse search system, such as the mass spectral Probability-Based Matching (Pesyna et al., 1976), select the search features to be retained from the library record using, almost exclusively, the inverse probability of occurrence of these features within the complete data base. It is assumed that the less frequently a feature is found in the whole data base, the more characteristic, and hence the more useful in searching it will be. The mass-to-charge values of the lines in low-resolution mass spectra are generally rounded to the nearest integral value for use by search algorithms. With powder diffraction patterns, however, the d values of the lines are measured on a continuous scale, and this complicates the process of selecting the lines to be used in a search system.
Both the continuous nature of the d-spacing values and the non-linearity of the experimental errors therein were accommodated in the present system by using, in the search file, rather than the d spacings themselves, values of d*, defined as 1000/d rounded to the nearest integer. This technique has been applied in previous powder-diffraction search systems (Johnson & Vand, 1967). If it is assumed (Vand, 1956) that experimental errors in d are proportional to the square of d, then it follows that the d* values should exhibit constant errors, independent of the actual d* values***.
The variations in frequency of occurrence of d* values that were encountered in the JCPDS data base appear not to be a function of the basic properties of the materials from which the data derived, as is the case, for example, for the distribution of m/e peak positions in a library of mass spectra. Rather, a raw plot of the occurrence frequencies of the integer-valued d* exhibits a cyclical fine structure that is evidently traceable to the nearest unit (or half-unit) on the measurement scale - in this case, to the nearest mm (or half mm) of circumferential film dimension. Furthermore, the relative scarcity of pattern lines at the smaller d spacings is presumed for the most part to be a manifestation of the experimental difficulties of reading lines at the corresponding extreme diffraction angles. This deficiency seems to be particularly pronounced in patterns that are in the earlier sets of the JCPDS data base and which, therefore, may have been measured as many as forty years ago. In assigning probabilistic weights to patterns lines for purposes of selecting the subset to be retained in the search file, the fine structure was ignored. The contributions from lines occurring infrequently at extreme d spacings are used, but an upper limit is placed upon their maximum contribution to the selection criterion, relative to the contributions due to the lines intensities.
This approach does not provide a complete solution to the problem because, given a pattern containing equally intense lines at 0-9 and 2-0A, that at 0-9 A represents a less frequently encountered d spacing and would be more likely to be selected for inclusion in the search file. This would have resulted in an unrealistic weighting of the search files in favor of the lines with low d values and, in such a case, should a user submit for identification a pattern containing only the more easily read lines with larger d spacings, no match would result, even though the submitted lines were both numerous and accurate. Inclusion in the search file records of a large number of lines is a feature, for example, of Johnson's (1977) algorithm. While this ensures the retention of the mid-range d spacings, it substantially increases the time and cost of the general search, and still leaves open the possibility of some library patterns so saturating their research records with small d spacings that the typical user would never obtain an identification for the phases they represent.
For these reasons, a two-part search file organization was devise. As will be seen, this
efficiently serves the user who does not or cannot observe the smaller d-spacing lines, while also
providing improved performance for the user who does record and enter these values. The
principal search file contains, in each of its records, the best 15 lines from the reference file at d
spacings larger than 1-20 A. These lines are selected on the basis of the intensity-weighted
inverse probability of their occurrence in the data base. For those reference patterns that contain
lines below this value, an entry is made in the principal search file record that directs the search
program to a secondary record, in another file, that contains the best eight lines below 1-20 A.
This secondary file is accessed if, and only if, te reference pattern actually contains such lines,
and the user's range of observation extends to below 1-20A.
(b) Organization of the search file
The editing of the reference file to produce a highly compact, efficiently organized search file
is the first of the steps used to achieve the efficiency of operation necessary for an effective and
economically viable interactive reverse search procedure. The ordering of the file is also
important, in that the search records that are retrieved should be closely clustered so as to
facilitate the retrieval. This requires dense blocking and packing of the search records, so that a
number of closely clustered candidates search records can be retrieved in a single mass-storage
access.
(c ) Prescreens
Prescreens are used to reduce the final number of candidate phases that need be submitted to the search/match algorithm. The prescreens that have been found to be effective in this sense include screens that can be controlled by the user and screens that are used automatically by the algorithm. The program, as an example, uses the stronger lines in the user's problem pattern to select candidate matching phases for further examination.
The user-controlled prescreens include the selection of substance classes (minerals, non-minerals, organic, inorganic, etc.), either singly or in combination, minimum acceptable reference pattern quality, or selection of alloys or non-alloys. The user can also select the JCPDS volume or set number if it is desired to restrict the search to some such subset of the whole data base.
The most powerful user-controlled prescreen is that based upon the elemental composition of the sample. Both expected and prohibited elements can be named, as well as expected or prohibited functional groups. A search may be chemically restricted, so that only the named elements are permitted. Alternatively, additional elements may be allowed, provided that at least one desired element and none of the prohibited elements are present. Functional groups can be handled in just the same way as elements.
Finally, a lower limit, in terms of atomic number, can be placed upon the elements allowed.
Elements with atomic number lower than this user-specified limit can occur without restriction.
In particular, phases containing only elements with atomic number below this limit will always
be examined.
(d) Presentation of results
When a library pattern is retrieved during a search, the maximum possible "similarity index"
(SI) between it and the unknown patters is calculated. This SI value is a measure of the
confidence of the match of the library pattern to the unknown data. The program then computes
the actual SI value for the library pattern, under the user-specified experimental conditions and
limitations, and finally the overall intensity of the image of that library pattern in the user's
pattern. This is reported to the user as %'. The calculation of the actual SI value involves
scaling of the entered pattern against the library pattern, and takes into account the user's
experimental conditions. For example, a reference pattern line that, after scaling, is below the
user's background intensity in not counted against the match, should it be missing form the
user's pattern. The same consideration hold true for reference lines outside the user's range of d
spacing observation. Similarly, the user may specify the accuracy of the submitted d spacings
and intensities; these values are used to establish tolerance Windows on both d spacing and
intensity, within which a corresponding reference pattern line is deemed to have been matched.
Finally, the program allows for the possibility that a reference pattern line may be present in the
user's pattern at far too strong an intensity, due to an overlapping line from another phase. The
items 'NL/ML/X' that may be seen in
Fig. 1 ,
2 and
4 refer respectively to the number of lines in
the library pattern that would survive any of the user-defined parameters, the number of lines
which were found in both the library pattern and the unknown pattern and the number of lines
that are in the library pattern but absent from the user's data.
Since the system is designed for interactive operation, it contains a number of auxiliary functions designed to assist a user in this environment. A request for 'help', typed just so, is honored virtually anywhere, and results in the user obtaining an informative message regarding the various options available at that particular point in the program, and the effects of each.
The diffraction pattern that constitutes the 'unknown' may be entered manually by the user. Alternatively, the user can take advantage on any of the computer system's utilities to create 'save' files of problem patterns directly from a suitably equipped or interfaced diffractometer, or from a paper tape or cassette prepared in an offline mode, and thus avoid manual input completely.
The diffraction pattern of the unknown is entered into the search programs beginning with two free-form title lines, followed by free-format (d, I) pairs, one per line. 20 values cannot currently be accepted by the program. When all the data have been entered, a blank line is entered to signify this fact. The user is then offered an opportunity to review and/or modify this pattern. In the modification process, which can be invoked at any point during the user session, the entry of a previously supplied d spacing replaces the earlier intensity at that d spacing with the newly entered value. If this new intensity is zero, or omitted, the line will be deleted from the pattern.
It is not required that the user pattern be normalized; the program will normalize the data before searching. However, the pattern is also saved exactly as entered, free of normalization or d-spacing quantization, for display and possibly further modification by the user. The title lines are also retained; should the user select to retain the pattern on a disk file for future reference, the title forms part of the saved information, for display to the user whenever this saved pattern should be retrieved.
Before beginning the search, the user may elect to limit the search to one or more of the various subfiles. Limitations based upon the presence or absence of elements may also be imposed at this point, or at any other time during the session.
The algorithm computes values for the background intensity and the d-spacing range from the user pattern; it then solicits alternative values from the user. It is particularly important that a reasonable upper limit upon the observable d-spacing range be supplied, so that incorrect potential matches might be discarded during the ensuing search. The values of the user-supplied d spacings are employed by the program to select the appropriate subset of the data base, prior to beginning the reverse search.
All of the user-submitted lines are used in the search. The numbers of those reference patterns that the search finds to be imbedded in the user patter are tabulated internally, with the top-ranked matches being listed, in descending order of match reliability, at the end of the search. At the user's option, the next-ranked batch of answers will be listed, also ordered, and so on, as far down the list of matching patterns as the user cares to go.
Each listed match includes the JCPDS pattern number, the measures of match reliability (the similarity index, the number of lines used and matched, etc.), and the molecular formula and name of the source material. Chemical Abstracts Service Registry Numbers are currently being added to the data base, as in the case for all CIS files (Heller et al.,1976). Currently, JCPDS sets 1 through 26 are undergoing CAS registration.
The pattern option of PDSM allows the user to view the complete record for any of the patterns in the JCPDS data base. In this display, the lines specifically selected for inclusion in the PDSM search process are highlighted. Additionally, the user may ask to have specific reference patterns tested against the problem pattern, without performing a complete search. In this way, preconceived possibilities may be quickly tested. More importantly, a user unsure of the measurement accuracies appropriate to the problem pattern can observe the effects of different d spacing and intensity error tolerances upon the goodness of fit of phases known to be present, and thereby select tolerance values suitable for a complete search.
The powder diffraction search-match system does not currently contain a procedure for the
automatic 'substraction' of identified phases from the unknown pattern. A program has been
developed which permits substraction of identified phases from the experimental data and a
repeat of the search through the residual data. This program is currently being tested in a private
version of PDSM and will be the subject of a subsequent paper.
In 1977 and 1978, a group at the National Bureau of Standards (NBS), in cooperation with the Computer Sub-Committee of the JCPDS designed, for the purposes of conducting a 'round robin' test of the search systems, a series of sets of X-ray powder diffraction data (Jenkins, 1977; Jenkins & Hubbard, 1979; Jenkins, Pearlman & Hahm, 1979). Each of the sets of data corresponded to a mixture of at least three phases. Some sets of phases were completely inorganic and others were composed only of organic materials.
The Powder Diffraction Search-Match system was tested with these data sets and scored well over 95% retrieval of the correct phases. In this section, two searches, selected from the round robin, are presented. The results obtained form the first of these are given in Fig. 1. The data provided were derived from four inorganic materials; ferric sulfide (greigite), copper sulfide (covellite), ferrous sulfide (pyrite) and copper iron oxide.
No prior knowledge regarding the elemental composition on any of the constituent phases was available and so the search was carried out without the benefit of any user imposed prescreens. As can be seen from Fig. 1 , the first eight phases identified by the program included the four correct phases (underlined). In addition, three of the four top-ranked phases were equivalent to one or other of the correct phases.
Fig. 1. Round robin test 1A. Mixture of Fe2CuO4, Fe3S4, CuS and FeS2
Fig. 2. Round robin test 6A. Mixture of m-dinitrobenzene,
2, 4-dinitrophenol and 2, 6-dinitrophenol.
In a second example, shown in
Fig. 2 , the pattern given by a
mixture of three organic
compounds, m-dinitrobenzene, 2,4-dinitrophenol and 2, 6-dinitrophenol was used as the
unknown data set. In this instance, the search was restricted to the organic subfile of the data
base, a group of 10 139 patterns. The three correct patterns were identified among the first five
answers, which also included an earlier pattern for the 2, 4-dinitrophenol and the pattern given by
D-arabinose.
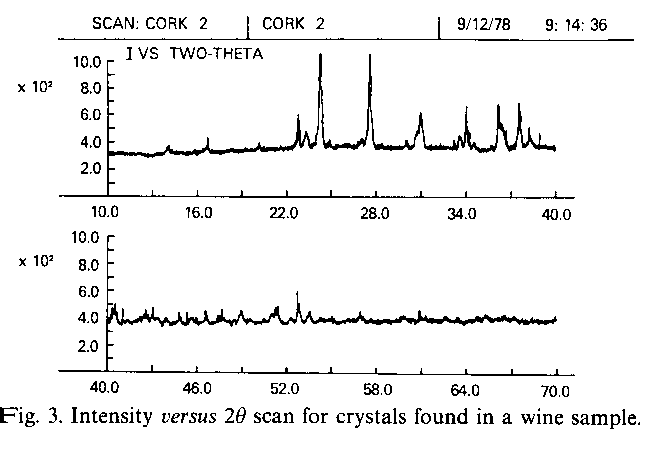
Fig. 3. Intensity versus 20 scan for crystals found in a wine sample.
Fig. 4. Search using the data from crystals found in a wine sample.
The quality of the data used in the round robin tests could be described as good, with Ad/d values of 3/1000 and AI/I of the order of 1/10. Results of previous round robins (Jenkins, 1977) have indicated that computer search-match programs are very sensitive to the Ad/d values for the data used.
When the quality of the experimental data is considerably poorer than in the round robin tests, the search-match programs still produce useful results for a tolerably small investment of time and money, as may be seen from a third example. When a bottle of modestly priced California Cabernet Sauvignon wine was uncorked, a number of small crystals were found adhering to the inside surface of the cork. The total weight of these crystals was about 50 mg. The crystals were scraped off the cork and ground in a mortar. About 20 mg of the resulting powder was spread on the surface of a glass slide using amyl acetate as a dispersant. The diffractogram shown in Fig. 3 was recorded between 10 and 70 20 in total time of 10 min, using an automated powder diffractometer Jenkins & Hubbard, 1979; Jenkins, Pearlman & Hahm, 1979). This diffractogram in not of a high quality because it was acquired relatively rapidly on a small amount of sample. Relatively few well defined peaks are seen and the signal to noise ratio is low. This example was chosen deliberately however, in an attempt to approximate the sorts of conditions that prevail in a typical routine measurement. A peak searching algorithm was used to locate the peak maxima and the d values were calculated from the 20's using Bragg law.
These data were then entered into the search-match system as shown in Fig. 4 . No elemental information was used at this stage and only the organic subfile was searched. As can be seen from Fig. 4 , this led to an examination of 777 patterns, of which 36 were considered to be possible solutions. The correct result in this case was potassium hydrogen tartrate, which was found as the top-ranked answer with a SI index of 110/151.
A repeat of the search after X-ray fluorescence analysis had revealed that potassium was a
major constituent of the material reduced the number of potential solutions to four. The correct
answer was the top ranked of these. The entire problem, excluding data acquisition, but
including data entry, was handled in an elapsed time of 12 min 16 s. The c. p. u. time was 17 s
and the overall computer charge was $13.00.
The major purpose in embarking upon this project was to design an interactive search system of programs that could be used successfully with the data base of X-ray powder diffraction patterns.
The programs described above appear to deal with the major technical problems inherent in such searching, and the remaining question concerns means whereby the system can be made widely available. To this end, the PDSM system has been merged into the NIH-EPA Chemical Information System, and the programs and data base have been installed in the DECsystem-10 computers of the Interactive Sciences Corporation. An annual subscription fee of $300 is required for access to the system. All searching is billed at the rate of $60 per wall-clock hour. Searches of the sort discussed above therefore cost between $5 and $20. Access to the computer system is by local telephone call, using the Telenet communications network in most cities in the US, and a toll free '800' long-distance telephone call elsewhere.
Those wishing further information regarding access to the system are invited to contact the
JCPDS or the CIS Operations manager (GWAM).
Abramson, F. P. (1975). Anal. Chem. 47, 45-49.
Dalrymple, D. L., Wilkins, C. L., Milne, G. W. A. & Heller, S. R. (1978). Org. Magn. Reson. 11, 535-540.
Frevel, L. K. (1965). Anal. Chem. 37, 471-482
Frevel, L. K. & Adams, C. E. (1968). Anal. Chem. 40, 1335-1340.
Frevel, L. K., Adams, C. E. & Ruhberg, L. R. (1976). J. Appl. Cryst. 9, 300-305.
Heller, S. R., Milne, G. W. A., Feldmann, R. J. & Heller, S. R. (1976). J. Chem, Inf. Comput. Sci. 16, 176-178.
Heller, S. R., Milne, G. W. A. & Feldmann, R. J. (1976). J. Chem. Inf. Comput. Sci. 16, 232- 233.
Heller, S. R., Milne, G. W. A. & Feldmann, R. J. (1977). Science, 195, 253-259.
Jenkins, R. (1977). Adv. X-ray Anal. 20, 125-137.
Jenkins, R. & Hubbard, C. (1979). Adv. X-ray Anal. 22, in the press.
Jenkins, R., Pearlman, S. & Hahm, Y. H. (1979). Norelco Rep. In the press.
Johnson, G. G. (1977). Resolution of X-Ray Power Patterns, edited by J. S. Mattson, H. B. Mark & H. C. MacDonald, Chapter 3, pp. 45-87. New York: M. Dekker & Sons.
Johnson, G. G. & Vand, V. (1967). Ind. Eng. Chem. 58, 19-31.
Nichols, M. C. (1966). Twenty fourth Pittsburgh Diffraction Conf., B-3.
Pesyna, G. M., Venkataraghavan, R., Dayringer, H. E. & McLafferty, F. W. (1976). Anal. Chem. 48, 1362-1368.
Van, V. (1956). Fourteenth Pittsburgh Diffraction Conf., Paper 8.
Footnotes:
* Toxic Substances Control Act of 1976, 15 U. S. C. 2601.
** The Joint Committee on Powder Diffraction Standards, 2601 Park Lane, Swarthmore, PA 19081, USA.
*** There is some discussion as to the adequacy of this coarse a quantization, since modern diffractometers are already capable of accuracies and resolution approaching or even exceeding the quantization error so obtained. The question to be settled, however, is not whether these d* values retain the implicit accuracy of such patterns; it is evident they do not. Rather, the concern should be the value of increasing the quantization accuracy to the intended function of PDMS, vis: the identification and ranking of candidate phases from a predetermined library of possibilities. This question will only be answered through the experiences encountered in using PDMS, and the identification of actual problems that were not satisfactorily treated solely because of excessive quantization error.
{kind=link}
{kind=link}
{kind=link}
{kind=link}